Tutorial on the usage of micelle_whole tool
Here two examples on how to use the make_micelle_whole tool for different polymers will be illustrated.
Before starting any analysis, load the neccesary modules for this class.
from utils_mda import MDA_input
#from pysoftk.pol_analysis.tools.utils_mda import MDA_input
from utils_tools import *
#from pysoftk.pol_analysis.tools.utils_tools import *
from clustering import SCP
#from pysoftk.pol_analysis.clustering import SCP
from make_micelle_whole import micelle_whole
#from pysoftk.pol_analysis.make_micelle_whole import micelle_whole
import numpy as np
import pandas as pd
/home/raquellrdc/Desktop/PhD/mda_umap/lib/python3.10/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
Obtain largest micelle
Here we will show how to obtain the largest micelle from the resutls obtained from the SCP class. This is convenient if you want to perform the analysis in the largest aggregate of the simulation
Select your trajectory files, it is recommended to use a tpr file for the topology and xtc file for the trajectory. Note that any MDAnalysis supported file can be used here.
topology='data/short_movie_cyclic.tpr'
trajectory='data/short_movie_cyclic.xtc'
Import the clustering data from SCP function
resids_total='data/pictures_tutorial/cyclic_scp_result.parquet'
Obtain the largest micelle from the clustering pandas dataframe
largest_micelle_resids = micelle_whole(topology, trajectory).obtain_largest_micelle_resids(resids_total)
‘largest_micelle_resids’ is a np.array with the resids of the molecules that belong to the same cluster of the steps of the trajectory where SCP was ran.
largest_micelle_resids
[array([ 1, 2, 3, 4, 6, 7, 8, 11, 12, 13, 14, 15, 17, 18, 19]),
array([ 2, 4, 6, 7, 13, 14, 15, 17, 18, 19, 20]),
array([ 2, 4, 6, 7, 13, 14, 15, 17, 18, 19, 20])]
Note that you you do not neccesarily need to only work with the largest micelle. The SCP tool ouputs all micelles of the system in a pandas dataframe so that you can select at each time step which ever ones you prefer. For example if you want to to take the two smalles micelles of the system you could do it in the following way
#load the dataframe
df_results = 'data/pictures_tutorial/cyclic_scp_result.parquet'
df = pd.read_parquet(df_results)
# Define a function to find the two smallest micelles (so more than 1 polymer)
def find_two_smallest_lists(lst):
sorted_lists = sorted(lst, key=len)
return sorted_lists[:2]
# Apply the function to each row in the DataFrame
df['smallest_lists'] = df['micelle_resids'].apply(find_two_smallest_lists)
#the following column contains the resids of the two smallest aggregates at each time step!
df['smallest_lists']
0 [[16, 9, 10, 5], [1, 2, 3, 4, 6, 7, 8, 11, 12,...
1 [[1, 3, 12], [5, 8, 9, 10, 16]]
2 [[1, 3, 5, 8, 10, 12, 16], [2, 4, 6, 7, 13, 14...
Name: smallest_lists, dtype: object
Obtain the whole coordinates of a molecular structure
Now, let’s obtain the coordinates of the largest micelle made whole across the pbc
Let’s define the resname of the molecules that we want to make whole. More than one resname can be inputted. Note that it should be the resname of the molecules of the largest_micelle array.
resname=['LIG']
Also, define the start, step and step of frames that you want to run the analysis on. Note that they need to be the same as the ones you ran the SCP clustering on.
start=0
stop=10001
step=1
Now, we are ready to obtain the whole coordinates of the micelle!
atom_pos = micelle_whole(topology, trajectory).running_make_cluster_whole(resname, largest_micelle_resids, start, stop, step)
0%| | 0/3 [00:00<?, ?it/s]/home/raquellrdc/Desktop/PhD/pysoftk/alejandro_newest_releast_check/pysoftk_analysis_code/test_final/make_micelle_whole.py:347: FutureWarning: arrays to stack must be passed as a "sequence" type such as list or tuple. Support for non-sequence iterables such as generators is deprecated as of NumPy 1.16 and will raise an error in the future.
atom_positions_over_trajectory = list(tqdm(map(self.make_cluster_whole, frames, resname, cluster_resids_f[0],
100%|████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 3/3 [00:00<00:00, 13.98it/s]
Elapsed time for matrix calculation: 0.3085 seconds
Now, atom_pos contains the coordinates of all the atoms of the micelle made whole at each time step selected. In each array, the first element is the time frame of the analysis, and the second the positions array
atom_pos
[(0,
array([[73.69 , 81.16 , 77.950005],
[73.450005, 81.69 , 77.04001 ],
[74.51 , 81.770004, 78.340004],
...,
[75.19 , 47.36 , 76.54 ],
[73.490005, 47.280003, 76.600006],
[72.19 , 46.860004, 74.130005]], dtype=float32)),
(1,
array([[ 16.060001 , 116.100006 , 3.3600001],
[ 17.04 , 116.14 , 3.8300002],
[ 15.860001 , 117.17001 , 3.3000002],
...,
[ 22.560001 , 98.76 , -1.7771301],
[ 22.380001 , 97.03 , -1.407135 ],
[ 22.480001 , 98.73 , 1.0200001]], dtype=float32)),
(2,
array([[ -7.5497437, -37.959747 , 111.810005 ],
[ -6.5297394, -37.979744 , 112.200005 ],
[ -7.419739 , -37.42974 , 110.87001 ],
...,
[ -2.0497437, -28.63974 , 115.00001 ],
[ -1.9997406, -27.959747 , 116.520004 ],
[ -3.739746 , -25.669746 , 116.490005 ]], dtype=float32))]
Obtain pdb file of the structure made whole
Using the ouputs from above, we can select the inputs that we need to obtain the whole snapshot. First, we need to define are the name of the output pdb and the frame at which you want to obtain the pdb. Note that the frame you selected will depend on which frames you ran the analysis. For example, if you ran the analysis on 3 frames, and you want to obtain the snapshot of the last frame, you will need to select frame at position 2, since this one will be the last one.
snapshot_frame=2
snapshot_name='data/pictures_tutorial/cyclic_micelle_whole.pdb'
Then, we also need to input the whole positions of the atoms, and their respective resids that we want in the snapshot. Since we want the whole micelle and frame 2, it is as easy as selecting item 2 from atom_pos (whole positions) and largest_micelle_resids (resids of polymers in the same cluster)
atom_pos_frame=atom_pos[2]
largest_micelle_resids_frame=largest_micelle_resids[2]
Now, we can obtain the snapshot
snapshot = micelle_whole(topology, trajectory).obtain_snapshot(snapshot_name, atom_pos_frame,
largest_micelle_resids_frame, resname, snapshot_frame)
3971
3971
/home/raquellrdc/Desktop/PhD/mda_umap/lib/python3.10/site-packages/MDAnalysis/coordinates/PDB.py:1151: UserWarning: Found no information for attr: 'altLocs' Using default value of ' '
warnings.warn("Found no information for attr: '{}'"
/home/raquellrdc/Desktop/PhD/mda_umap/lib/python3.10/site-packages/MDAnalysis/coordinates/PDB.py:1151: UserWarning: Found no information for attr: 'icodes' Using default value of ' '
warnings.warn("Found no information for attr: '{}'"
/home/raquellrdc/Desktop/PhD/mda_umap/lib/python3.10/site-packages/MDAnalysis/coordinates/PDB.py:1151: UserWarning: Found no information for attr: 'chainIDs' Using default value of ''
warnings.warn("Found no information for attr: '{}'"
/home/raquellrdc/Desktop/PhD/mda_umap/lib/python3.10/site-packages/MDAnalysis/coordinates/PDB.py:1151: UserWarning: Found no information for attr: 'occupancies' Using default value of '1.0'
warnings.warn("Found no information for attr: '{}'"
/home/raquellrdc/Desktop/PhD/mda_umap/lib/python3.10/site-packages/MDAnalysis/coordinates/PDB.py:1151: UserWarning: Found no information for attr: 'tempfactors' Using default value of '0.0'
warnings.warn("Found no information for attr: '{}'"
/home/raquellrdc/Desktop/PhD/mda_umap/lib/python3.10/site-packages/MDAnalysis/coordinates/PDB.py:1151: UserWarning: Found no information for attr: 'elements' Using default value of ' '
warnings.warn("Found no information for attr: '{}'"
/home/raquellrdc/Desktop/PhD/mda_umap/lib/python3.10/site-packages/MDAnalysis/coordinates/PDB.py:1151: UserWarning: Found no information for attr: 'record_types' Using default value of 'ATOM'
warnings.warn("Found no information for attr: '{}'"
/home/raquellrdc/Desktop/PhD/mda_umap/lib/python3.10/site-packages/MDAnalysis/coordinates/PDB.py:1151: UserWarning: Found no information for attr: 'formalcharges' Using default value of '0'
warnings.warn("Found no information for attr: '{}'"
/home/raquellrdc/Desktop/PhD/mda_umap/lib/python3.10/site-packages/MDAnalysis/coordinates/PDB.py:1198: UserWarning: Found missing chainIDs. Corresponding atoms will use value of 'X'
warnings.warn("Found missing chainIDs."
/home/raquellrdc/Desktop/PhD/mda_umap/lib/python3.10/site-packages/MDAnalysis/topology/PDBParser.py:331: UserWarning: Element information is missing, elements attribute will not be populated. If needed these can be guessed using MDAnalysis.topology.guessers.
warnings.warn("Element information is missing, elements attribute "
The visualization on VMD of this step of the trajectory before applying make_micelle_whole is:

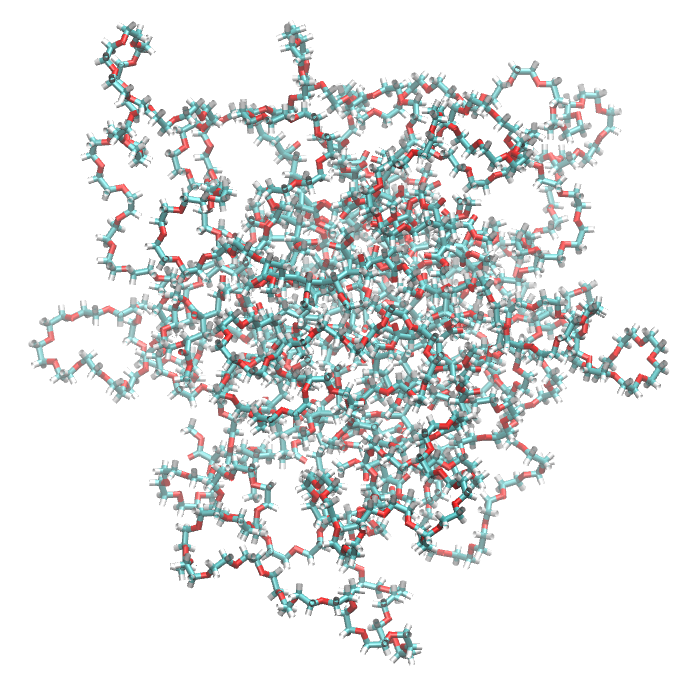
This is the visualizaiton on VMD of the pdb file produced with make_micelle_whole. It has made it perfectly whole !